Лиссэнцефалия что это такое
Узловые гетеротопии серого вещества присутствуют у многих пациентов с другими нарушениями миграции, такими как полимикрогирия или шизэнцефалия. Одиночные или многоочаговые крупные гетеротопические узлы могут являться фокусами парциальных судорог. Однако даже гигантские гетеротопии, затрагивающие одно полушарие, могут оставаться бессимптомными. Детализированное нейропсихологическое исследование одного из таких случаев продемонстрировало едва уловимые нарушения полушарных функций, несмотря на нормально развитый интеллект (Calabrese et al., 1994).
Спектр классической лиссэнцефалии и субкортикальной линейной гетеротопии. Эти нарушения миграции могут быть рассмотрены для оценки различных степеней тяжести основной патологии нейрональной миграции, хотя генетически они различаются (Palmini et al., 1993).
Под лиссэнцефалией понимают гладкий мозг. Термин агирия-пахигирия лучше, так как поверхность мозга не всегда гладкая (Aicardi, 1991). В наиболее тяжелых случаях извилины не формируются (агирия). В большинстве случаев присутствует несколько извилин (пахигирия). Dobyns и Leventer (2003) различают 6 степеней лиссэнцефалии (от 1 до 6), в зависимости от количества извилин, видимых на МРТ. Только степень I заслуживает названия лиссэнцефалии; степени 2-4 являются случаями с пахигирией, и степени 5 и 6 относятся к субкортикальной линейной гетеротопии. В данном разделе объединены различные типы, как имеющие сходный спектр и, очевидно, отчасти сходные механизмы. Хотя имеется несколько форм лиссэнцефалии, в этом разделе рассмотрен только вариант мутации гена LIS1 на 17 хромосоме.
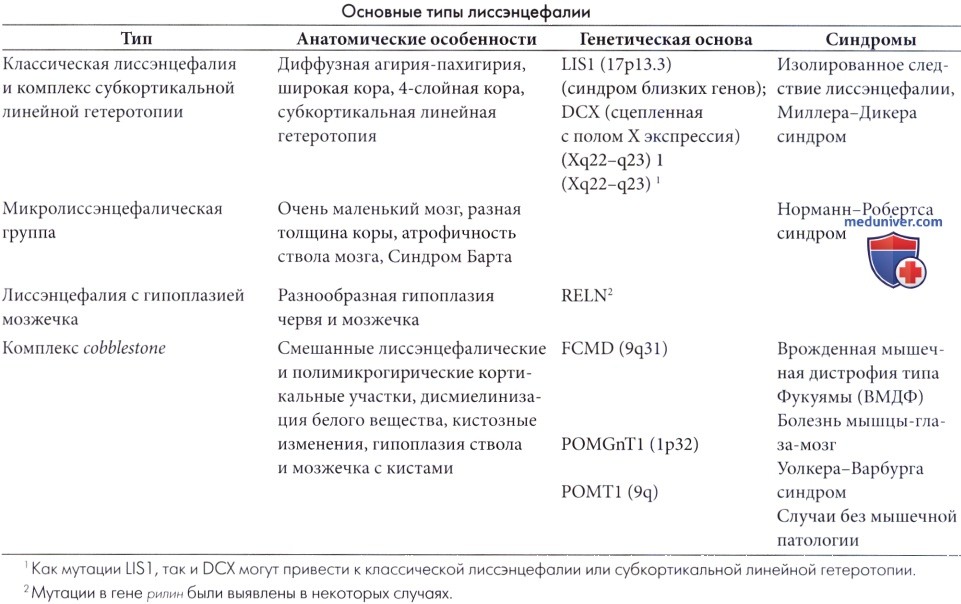
• Классическая (тип 1, Бильшовского) лиссэнцефалия. При классической лиссэнцефалии мозг имеет малые размеры и только первичные, и иногда несколько вторичных извилин. При отсутствии извилин извилистыми становятся сосуды. Кора патологически утолщена (10-20 мм), тогда как белое вещество выглядит узкой полосой вдоль желудочков. Типично наличие четырех слоев коры:
1) поверхностный, разреженный клеточный слой, аналогичный молекулярному слою нормального мозга;
2) узкий, густоклеточный слой, где располагаются большие пирамидальные нейроны, которые в норме должны располагаться в более глубоких отделах;
3) тонкий слой белого вещества, ниже которого находится
4) широкая полоса малых эктопированных нейронов, распрострающаяся почти до стенки желудочков (Dobyns и Leventer, 2003).
Многие нейроны в клеточных слоях ориентированы неправильно, с апикальным дендритом, направленным вниз или вбок (Takashima et al., 1987). Более глубокий клеточный слой сформирован из эктопированных нейронов, остановившихся на пути их миграции из герминативного слоя к коре примерно на 12 неделе гестации, поэтому кора выглядит как у 13-недельного плода. Нейроны этого слоя имеют избыточную колонковую организацию. В продолговатом мозге характерна эктопия ядра оливы. Зубчатые ядра ненормально запутанны, и пирамиды гипоплазированы или отсутствуют (Friede, 1989).
Агенезия мозолистого тела при таком типе необычна. Тип I лиссэнцефалии в 65% случаев возникает в результате мутации гена LIS1, который кодирует 46D белок, некаталитическую часть ацетилгидролазы фактора активации тромбоцитов (Bix и Clark, 1998, Gleeson et al., 1999). Большинство случаев носит спорадический характер. Зарегистрированы случаи в связи с врожденной цитомегаловирусной инфекцией, но с разными патологическим изменениями (Hayward et al., 1991). Часть случаев возникает в связи с хромосомной патологией, делецией дистальной части короткого плеча 17 хромосомы (17р13.3).
Некоторые из таких случаев являются частью специфического дисморфического близкого генного синдрома, синдрома Миллера-Дикера, который характеризуется узким лбом, широкой переносицей, отсутствием выемки верхней губы, вздернутыми ноздрями, ретрогнатизмом, аномалиями пальцев и гиперваскуляризацией сетчатки (Dobyns и Leventer, 2003). В таких случаях Dobyns и Truwit (1995) выявили явную делецию 17р13.3 у 14 из 25 пациентов и субмикроскопические делеции в 25 из 38 случаев с использованием цитогенетических методов и в 35 из 38 случаев с флюоресцентной гибридизацией in situ. Сиблинги с синдромом Миллера-Дикера рождались у пар, в которых у одного из родителей произошла пропорциональная транслокация концевого фрагмента хромосомы 17р на хромосому 13-15 пары, что проявлялось в несбалансированных формах у пострадавших детей (Greenberg et al., 1986, Dobyns и Leventer, 2003).
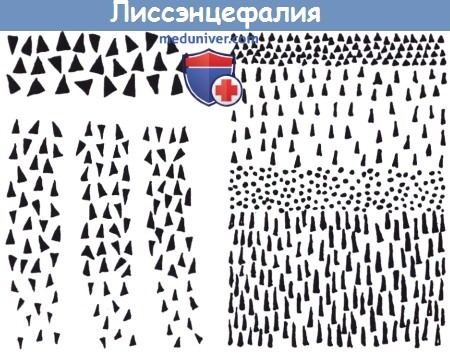 (слева) Тип I (классическая) лиссэнцефалия. Четырехслойная кора. От поверхности (сверху) вниз:
(слева) Тип I (классическая) лиссэнцефалия. Четырехслойная кора. От поверхности (сверху) вниз:
(1) молекулярный слой;
(2) поверхностный клеточный слой, содержащий несколько типов клеток, включая большие пирамиды, в норме располагающиеся глубже в пятом слое;
(3) широкий, бесклеточный слой;
(4) широкая полоса гетеротопированных клеток, остановленных при миграции — обратите внимание на столбчатое расположение.
(справа) Нормальное расположение.
Большинство случаев с типом I лиссэнцефалии не являются частью синдрома Миллера-Дикера и определяются как «изолированное» последствие лиссэнцефалии.
Клинические проявления во всех случаях отличаются тяжелой задержкой умственного развития и диплегией, часто атонического типа (de Rijk-van Andel et al., 1990). Как правило, имеются парциальные судороги и, как правило, инфантильные спазмы. У большинства пациентов присутствует некоторая степень микроцефалии, обычно легкой. При нехромосомной патологии дисморфизм не выражен, хотя лоб узкий и часто присутствует ретрогнатизм. Прогноз неблагоприятный, с ограниченной выживаемостью.
Некоторые случаи мутации LIS1 могут быть в большей степени связаны с субкортикальными групповыми гетеротопиями, нежели чем с лиссэнцефалией (Gleeson et al., 2000).
Диагноз типа I лиссэнцефалии стал возможным при помощи современных методов нейровизуализации. КТ и МРТ демонстрируют характерный внешний вид широкой кортикальной пластинки, с несколькими присутствующими или отсутствующими извилинами, отделенными от гиподенсивного белого вещества слегка волнистой или почти прямолинейной границей. Слоистость коры может быть выявлена при КТ или МРТ с высокой степенью разрешения. Патологические изменения обычно доминируют в задней части коры, в то время как несколько изгибов можно обнаружить спереди. При ультрасонографии уже с 18,5-25 недели определяется гладкость коры плода или новорожденного (Toi et al., 2004). МРТ дает более точные результаты (Ghai et al., 2006).
На ЭЭГ в большинстве случаев можно увидеть высокоамплитудную быструю активность альфа и бета частот, чередующихся даже на той же записи с высокоамплитудными дельта или тета медленными ритмами, которые могут имитировать медленные комплексы спайк-волн или гипсаритмию (de Rijk-van Andel et al., 1992, Quirk et al., 1993, Mori et al., 1994).
Дифференциальную диагностику проводят с другими состояниями, при которых имеется утолщение коры и нарушение послойного строения. Пахигирия в результате мутации LIS1 считается лишь легкой степенью лиссэнцефалии, не имеющей отношения к дифференциальному диагнозу. Определенные нарушения развития плода, особенно цитомегаловирусная инфекция, по-видимому, могут вызывать развитие пахигирии, гистологически связанной с полимикрогирией. Перивентрикулярная кальцификация может сопровождаться патологией формирования извилин мозга. В таких случаях микроскладки могут сливаться и походить на пахигирию.
Пренатальный диагноз не представляется возможным на поздних сроках беременности с помощью ультрасонографии, поскольку в это время только появляются третичные борозды (Toi et al., 2004). Исследования ДНК могут выявить мутировавший или отсутствующий ген LIS1. Для определения риска рецидива при поиске ламинарных гетеропий необходимы хромосомный анализ и МРТ родителей (особенно матерей).
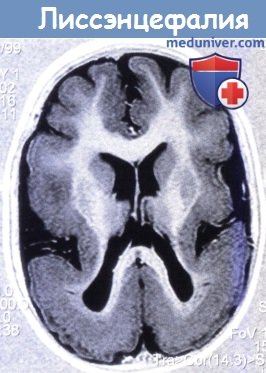 Классическая (тип I) лиссэнцефалия.
Классическая (тип I) лиссэнцефалия.
Т1-взвешенная аксиальная МРТ: (мутация LIS I) толстая корковая лента с гладкой поверхностью и прямой, неволнистой границей между серым и белым веществом.
Обратите внимание на присутствие нескольких мелких борозд в лобной области и полное отсутствие борозд сзади,
отсутствие оперкуляции с широко открытой сильвиевой бороздой и слоистость коры со слабой границей между гетеротопированными и полностью мигрировавшими нейронами.
• Субкортикальная ламинарная гетеротопия и лиссэнцефалия в результате мутации DCX гена. Ленточные гетеротопии (Barkovich et al., 1994, Franzoni et al., 1995) или «двойная кора» (Livingston и Aicardi, 1990, Palmini et al., 1991) являются результатом нарушенной миграции, при которой поверхностная кора, внешне нормальная или с отклонениями в извилинах, отделена тонким слоем белого вещества от полосы серого вещества. Граница между серым веществом и подлежащим белым веществом ровная как при агирии-пахигирии. Пациенты с этой аномалией часто страдают судорогами, которые могут иметь очаговый или генерализованный характер, иногда в форме синдрома Ленокса-Гасто и аномальной ЭЭГ (Hashimoto et al., 1993, Parmeggiani et al., 1994).
Нарушения умственного развития значительно варьируют, некоторые пациенты развиваются нормально (Livingston и Aicardi, 1990, Ianetti et al., 1993). Barkovich et al. (1994) при детальном изучении 27 случаев обнаружили значительную корреляцию между интеллектуальным уровнем и толщиной гетеротопической полосы; внешне нормальная кора была связана с лучшим развитием, но, вероятно, этот признак может варьировать. На ЭЭГ было обнаружено, что полоса способна продуцировать пароксизмальную активность и повышенный кровоток, как было продемонстрировано с помощью ОФЭКТ, что указывает на активацию коры.
Это состояние в большинстве случаев обусловлено сцепленной с полом мутацией DCX гена, кодирующего даблкортин (англ, doublecortin) (des Portes et al, 1998, Gleeson et al., 1999). Тем не менее, сходная мутация у мальчиков может привести к классической лиссэнцефалии (Pilz et al., 1998). Мутация очень разнообразно выражается у женщин и даже у некоторых мужчин (Cardoso et al. 2000, Gleeson 2000) и поэтому трудно распознается. Поэтому генетическое консультирование семьи, где родился мальчик с лиссэнцефалией, должно включать тщательный поиск ламинарной гетеротопии на МРТ и, если необходимо, мутации DCX у матери и сестер. Известны семьи, где пораженная мать рождала мальчиков с лиссэнцефалией и девочек с ламинарными гетеротопиями (Pinard et al., 1994). Редкие случаи ламинарной гетеротопии связаны с миссенс-мутацией LIS1 и с более умеренным фенотипом (Leventer et al., 2001).
При визуализации, тяжесть экспрессии у девочек варьирует от широких субкортикальных полос, иногда покрытых патологической корой, до с трудом обнаруживаемых тонких полос, которые видны только под ограниченными участками коры. Односторонние и частичные линейные гетеротопии иногда сложно распознать, для выявления могут потребоваться специальные срезы и изменение формата MPT (Gallucci et al., 1991). У мальчиков картина классической лиссэнцефалии такая же, как при LIS1. Однако передние отделы коры имеют более гладкую поверхность в сравнении с задними, в отличие от происходящего при мутации LIS1.
Эпилепсия, связанная с ламинарными гетеротопиями, может поддаваться медикаментозному лечению, но бывает и устойчивой. Хирургическое лечение оказалось неэффективным.
Синдром Барайтсера-Уинтера включает дисмор-фические признаки и пороки развития мозга в виде классической лиссэнцефалии или субкортикальных ламинарных гетеротопий (Rossi et al., 2003).
• Пахигирия. Этот тип представляет менее тяжелую форму спектра лиссэнцефалии и, вероятно, возникает в результате тех же механизмов. Однако эта форма гетерогенна и может входить в состав разных синдромов. Клинически пахигирия представлена различными схожими симптомами, но с меньшей тяжестью. На МРТ выявляют утолщение коры и линейное разделение между корой и белым веществом.
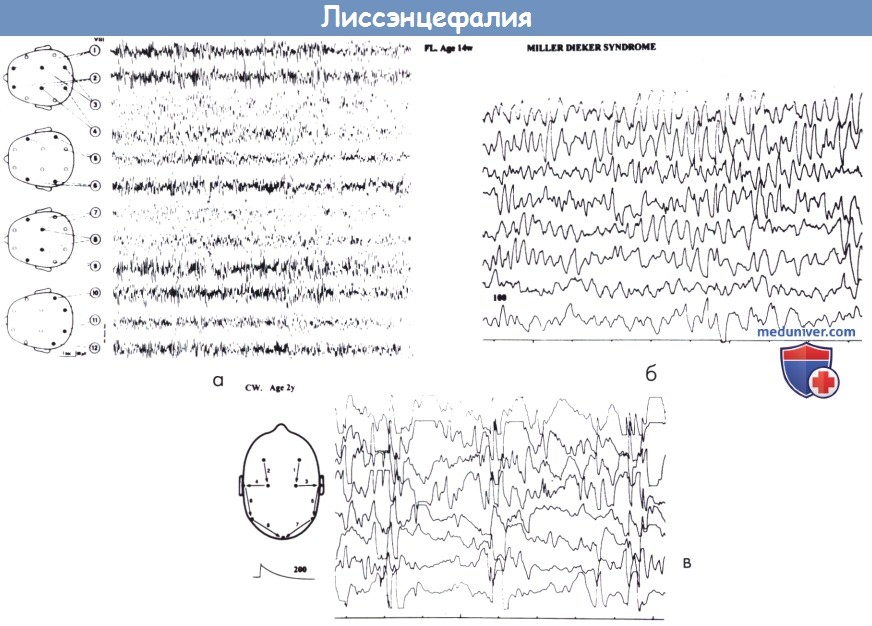 (а) Лиссэнцефалия-пахигирия у двухлетней девочки: ЭЭГ указывает на типичные быстрые ритмы с альфа и выше частотой.
(а) Лиссэнцефалия-пахигирия у двухлетней девочки: ЭЭГ указывает на типичные быстрые ритмы с альфа и выше частотой.
(б| Синдром Миллера-Дикера у 14-недельной девочки: ритмическая активность различных частот, но в основном в тета-диапазоне.
(в) Синдром Миллера-Дикера у двухлетнего мальчика: хотя присутствует некоторая избыточная тета-альфа активность, в записи преобладают повторяющиеся вспышки острых волн, достигающих 500-600 μВт.
• Другие формы и синдромы лиссэнцефалии. Распознание некоторых менее распространенных вариантов лиссэнцефалии не менее важно из-за разницы генетических и прогностических последствий (Hennekam и Barth, 2003, Raoul et al., 2003).
Микролиссэнцефалия состоит из крайне выраженной врожденной микроцефалии и агирии или пахигирии с широкой корой. Описано по меньшей мере, пять или шесть типов, передающихся по рецессивному типу, с различной степенью утолщения кортикального слоя, расположением имеющихся борозд и наличием сопутствующих пороков, таких как гипоплазия мозжечка, стволовая атрофия и увеличение желудочков (Ross et al., 2002, Dobyns и Leventer, 2003, Sztriha et al., 2004). Некоторые авторы (Dobyns и Barkovich, 1999) выделили эти случаи из «олигирической микроцефалии» (Hanefeld, 1999), которую они расценивают скорее как форму первичной микроцефалии, нежели форму расстройства миграции. Один из этих синдромов может быть связан с мутацией рилин гена (Hong et al., 2000, Crino, 2001).
Лиссэнцефалия с гипоплазией мозжечка является отдаленным проявлением микроцефалии с рудиментарной двуслойной корой мозга и тяжелой гипоплазией мозжечка (Ross et al., 2001, Sztriah et al., 2005). Вероятно, с рецессивным наследованием.
Лиссэнцефалия с гипоплазией мозолистого тела генетически гетерогенна. Некоторые случаи могут входить в группу мутации LIS1 или микролиссэнцефалии.
Х-сцепленная лиссэнцефалия с аномалией гениталий (XLAG) — врожденный порок с микроцефалией, тяжелой задержкой развития, тенденцией к гипотермии, отсутствием мозолистого тела и множественными аномалиями мозга (Berry-Kravis и Israel, 1994, Dobyns et al., 1999). Более вероятна гипоплазия гениталий, чем агенезия. XLAG развивается в результате мутации гомеобоксного гена ARX на хромосоме Х33.2 (Uyanik et al., 2003), на которой другие мутации также могут быть причиной некоторых неврологических синдромов (Kato et al. 2004, Suri 2005), включая Х-сцепленную задержку умственного развития (MRX54), агенезию мозолистого тела с патологией гениталий и синдром Партингтона с умственной отсталостью, атаксией и дистонией в зависимости от типа мутации.
Интересно, что лиссэнцефалия с неонатальными судорогами и тяжелыми аномалиями развития нервной системы, как было выявлено, связана с отсутствием глутамина.
Редактор: Искандер Милевски. Дата публикации: 29.11.2018
Лечение лиссэнцефалии в Израиле — полное купирование симптоматики
 Израильские специалисты по борьбе с врожденными аномалиями нервной системы хорошо известны во всем мире. Так как лиссэнцефалия связана с целым комплексом неврологических нарушений, в лечении пациентов принимает участие большая команда врачей. При этом диагностика занимает всего несколько дней.
Израильские специалисты по борьбе с врожденными аномалиями нервной системы хорошо известны во всем мире. Так как лиссэнцефалия связана с целым комплексом неврологических нарушений, в лечении пациентов принимает участие большая команда врачей. При этом диагностика занимает всего несколько дней.
В Топ Ихилов для терапии лиссэнцефалии применяются современные методы как консервативного, так и хирургического характера. Эпилептические приступы здесь предотвращают с помощью новейших антиконвульсантов, для лечения гидроцефалии проводят шунтирование мозга и эндоскопические вмешательства. В зависимости от индивидуальной клинической картины, пациент может проходить процедуры, направленные на коррекцию нарушенных функций дыхательной, выделительной и сердечно-сосудистой систем, опорно-двигательного аппарата.
Методы лечения лиссэнцефалии в Израиле
Лиссэнцефалия — редкое комплексное нарушение функций головного мозга, которое вызывается аномалиями нейрогенеза у плода в период с 21 по 24 неделю беременности. Большая часть нейронов не может достичь внешней части коры головного мозга и остается под кортикальной пластинкой. Как результат аномального развития, головной мозг новорожденного практически полностью лишен извилин и имеет гладкую поверхность. Дети с такой патологией обычно значительно отстают в развитии, но все определяется в каждом конкретном случае индивидуально.
Сегодня классифицировано более 20 различных видов лиссенцефалии, которые различаются клиническими проявлениями и особенностями мутации генов. Методы лечения лиссэнцефалии в Израиле опираются на купирование симптоматических проявлений, к которым относятся припадки, нарушения дыхательной функции, спастичность, трудности глотания и др.
В типичном случае пациент получает комплексное лечение. В состав команды врачей могут входить неврологи и педиатры, неонатологи и генетики, другие специалисты.
От эпилептических припадков страдает примерно 90% детей с лиссэнцефалией. Для их предупреждения пациенты получают лекарственные препараты из группы антиконвульсантов. Сегодня разработано множество эффективных средств — это некоторые транквилизаторы и синтетические аналоги ГАМК, производные янтарной кислоты и др. Особенно перспективными являются антиконвульсанты из группы аналогов ГАМК — гамма-аминомасляной кислоты, нейромедиатора, участвующего во многих процессах, происходящих в нервной системе. Современные аналоги ГАМК способны не только предотвращать эпилептические припадки, но и стабилизировать настроение пациента, снимать спастичность, снижать тревожность и бороться с депрессией.
Иногда у пациентов с лиссэнцефалией диагностируется водянка головного мозга. Для ее лечения могут проводиться различные виды шунтирования — создания искусственных путей для отвода излишков цереброспинальной жидкости из желудочков мозга в брюшную или грудную полость. Также широко применяются эндоскопические процедуры — вентрикулоцистерностомия, акведуктопластика и др.
При наличии проблем с глотанием пациенту может проводиться гастростомия — установка системы, позволяющей вводить пищу в желудок, минуя пищевод. Специалисты клиники владеют несколькими техниками проведения гастростомии, в том числе чрескожной эндоскопической, при которой вместо разреза брюшной стенки делается прокол, и весь необходимый микрохирургический инструмент вводится через него.
Для предотвращения мышечной дистрофии могут назначаться физиотерапия и лечебная физкультура.
Также пациент, в зависимости от индивидуальной клинической картины, может проходить лечение, направленное на купирование негативных проявлений со стороны сердечно-сосудистой, мочевыводящей, дыхательной систем и т. д.
Диагностика лиссэнцефалии в Израиле
Диагностика лиссэнцефалии в Израиле длится не более трех-четырех дней и проводится с помощью современного, сверхточного оборудования.
Первый день — консультация
Вместе с куратором-переводчиком пациент и сопровождающие отправляются в клинику на консультацию к ведущему специалисту.
Второй день — исследования
Далее проходят диагностические мероприятия.
Третий день — консилиум
Завершает программу исследований консилиум, в котором участвует сразу несколько профильных специалистов разных направлений — генетики, нейрохирурги, неврологи и т. д. Сообща они определяют вид лиссэнцефалии и планируют ряд лечебных мероприятий.
Лечение лиссэнцефалии в Израиле — стоимость
Сегодня многие предпочитают проходить лечение лиссэнцефалии в Израиле – цены на медицинские процедуры здесь гораздо более демократичны, чем в клиниках Соединенных Штатов, Австрии, Швеции или Франции. Прекрасное доказательство того, насколько приемлемо по стоимости лечение лиссэнцефалии в Израиле — отзывы таких людей. В них подчеркивается не только высокое качество лечебных процедур, но и их доступность.
Миллера-Дикера синдром
OMIM 247200
Наша команда профессионалов ответит на ваши вопросы
Синдром Миллера-Дикера (синдром лиссэнцефалии Миллера-Дикера OMIM 247200) – аутосомно-доминантное заболевание, обусловленное делецией генов локуса 17p13.3 (размер критического региона около 350-400 килобаз). Делеция локуса 17p13.3 приводит к нарушению миграции нейронов и в основном возникает de novo (образуется кольцевая хромосома, либо происходит делеция концевого участка короткого плеча), однако она может наследоваться от родителя со сбалансированной реципрокной транслокацией. Делеции всего гена PAFAH1B1 (другое название LIS1), лежащего в этом локусе, его отдельных экзонов и точковые мутации в нем приводят к развитию изолированной лиссэнцефалии. Формирование специфического фенотипа и более тяжелого течения заболевания при синдроме Миллера-Дикера связано с делецией более протяженного участка хромосомы, захватывающей несколько генов, в частности ген YWHAE, лежащий рядом с геном PAFAH1B1. Описаны случаи делеции только гена YWHAE. При этом у больных формируется фенотип, как при синдроме Миллера-Дикера (задержка роста, черепно-лицевые аномалии, задержка умственного развития), однако наблюдаются иные аномалии развития мозга, среди которых наиболее частыми являются расширение пространств Вирхова-Робина, мальформация Киари I, аномалии мозолистого тела. Описано также несколько случаев дупликации локуса 17p13.3, что также приводило к развитию синдрома Миллера-Дикера. Популяционная частота заболевания неизвестна.
В Центре Молекулярной Генетики методом количественной MLPA проводится поиск делеции / дупликации локуса локуса 17p13.3 и прямое автоматическое секвенирование гена PAFAH1B1.
Лиссэнцефалия головного мозга

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.

Среди органических церебральных патологий выделяется такая врожденная аномалия развития головного мозга, как лиссэнцефалия, суть которой заключается в почти гладкой поверхности коры его полушарий – с недостаточным количеством извилин и борозд. [1]
При полном отсутствии извилин определяется агирия, а наличие нескольких широких плоских извилин называют пахигирией. Данные пороки, как и некоторые другие редукционные деформации мозга, в МКБ-10 имеют код Q04.3.
Код по МКБ-10
Эпидемиология
Согласно статистике редких заболеваний, на 100 тыс. новорожденных приходится 1-1,2 случая лиссэнцефалии. [2], [3]
По некоторым данным, до 25-30% случаев классической лиссэнцефалии наблюдается у детей с синдромом Миллера-Дикера; почти у 85% пациентов выявляются точечные мутации и делеции генов LIS1 и DCX. [4]
Генетические исследования 17 генов, связанных с лиссэнцефалией, показали, что мутация или делеция LIS1 составляет 40% пациентов, а 23% связаны с мутацией DCX, за которыми следуют TUBA1A (5%) и DYNC1H1 (3%). [5]
Причины лиссэнцефалии
Все известные причины формирования коры мозга (cortex cerebri) почти или полностью без извилин и борозд, увеличивающих «рабочую зону» человеческого мозга и обеспечивающих «производительность» центральной нервной системы, связаны с нарушениями ее перинатального развития. То есть, развивается лиссэнцефалия у плода. [6]
Сбой в образовании слоев коры больших полушарий мозга плода при лиссэнцефалии – результат аномальной миграции образующих ее нейронов или преждевременной остановки данного процесса.
Этот важнейший для цереброкортикального гистогенеза процесс происходит в несколько этапов с 7 по 18 неделю беременности. И, учитывая его повышенную чувствительность к генетическим мутациям, а также различным негативным физическим, химическим и биологическим воздействиям, любое отклонение от нормы может привести к с неправильной локализации нейронов с возможным образованием утолщенного слоя серого вещества коры без характерной структуры. [7]
В некоторых случаях лиссэнцефалия у детей ассоциирована с синдромами Миллера-Дикера, Уокера-Варбурга или Нормана-Робертса.
Факторы риска
Патогенез
Не все случаи лиссэнцефалии имеют патогенез, обусловленный хромосомными аномалиями и генными мутациями. Но известны некоторые гены, которые кодируют белки, играющие важную роль в правильном перемещении нейробластов и нейронов вдоль клеток радиальной глии – для формирования коры головного мозга. И мутации этих генов приводят к данной патологии. [10]
В частности, это спорадические мутации (без наследственности) гена LIS1 на хромосоме 17, регулирующего цитоплазматический моторный белок микротрубочек динеин, а также гена DCX на X-хромосоме, кодирующего белок даблкортин (лиссэнцефалин-X). [11], В первом случае специалисты определяют классическую лиссэнцефалию (I типа), во втором – Х-сцепленную. [12]
При делеции гена FLN1, который кодирует фосфопротеин филамин 1, процесс направленной миграции нейронов вообще может не начаться, приводя к полному отсутствию извилин (агирии). [13]
Выявлены мутации гена CDK5, кодирующего киназный фермент – катализатор внутриклеточного обмена веществ, регулирующий клеточный цикл в нейронах ЦНС и обеспечивающий их нормальную миграцию в период пренатального формирования структур головного мозга.
Аномальные изменения гена RELN на хромосоме 7, вызывающие дефекты извилин коры больших полушарий при синдроме Нормана-Робертса, приводят в недостатку внеклеточного гликопротеина рилина, который необходим для упорядочивания миграции и позиционирования нервных стволовых клеток в ходе развития cortex cerebri. [14], [15], [16]
Ген ARX кодирует гомеобоксный белок, не имеющий отношения к aristalens, который является фактором транскрипции, играющим важную роль в переднем мозге и других тканях. [17] У детей с мутацией ARX наблюдаются другие симптомы, такие как отсутствие частей мозга (агенезия мозолистого тела), аномальные гениталии и тяжелая эпилепсия. [18], [19]
Цитомегаловирус (ЦМВ) связан с развитием лиссэнцефалии из-за снижения кровоснабжения головного мозга плода. Тяжесть ЦМВ-инфекции зависит от срока беременности. Раннее инфицирование с большей вероятностью вызовет лиссэнцефалию, поскольку миграция нейронов происходит на ранних сроках беременности. [21]
Кроме того, механизм возникновения данной аномалии включает неполную или более позднюю остановку перемещения нейронов из перивентрикулярной генеративной зоны в кору головного мозга. И в таких случаях развивается или неполная лиссэнцефалия, или пахигирия, при которой образуется несколько широких бороздок и извилин (но большая их часть отсутствует).
Симптомы лиссэнцефалии
Первые признаки данной патологии (при отсутствии ранее названных синдромов) могут проявиться не сразу после рождения, а спустя полтора-два месяца. И чаще всего наблюдаются такие клинические симптомы лиссэнцефалии, как:
Проблемы с глотанием обусловливают трудности с кормлением младенца. [22]
Высокая степень нейромоторных нарушений нередко проявляется тетраплегией – параличом всех конечностей. Возможна деформация рук, пальцев рук или ног.
При синдроме Нормана-Робертса с лиссэнцефалией I типа отмечаются черепно-лицевые аномалии: тяжелая микроцефалия, низкий наклон лба и выступающая широкая переносица, широко поставленные глаза (гипертерлоризм), недоразвитие челюстей (микрогнатия). [23]
При синдроме Миллера-Дикера также может быть аномально маленький размер головы с широким высоким лбом и коротким носом, углубления на висках (битемпоральные впадины), низко посаженные деформированные уши.
Синдром лиссэнцефалии тяжелой степени характеризуется микроцефалией, уменьшением размера глазных яблок (микрофтальмией) в сочетании с дисплазией сетчатки, обструктивной гидроцефалией и отсутствием или гипоплазией мозолистого тела.
Осложнения и последствия
Среди осложнений данной аномалии специалисты называют нарушение функции глотания (дисфагию) и гастроэзофагеальный рефлюкс; рефрактерную (неконтролируемую) эпилепсию; частые инфекции верхних дыхательных путей; пневмонию (включая хроническую аспирационную).
Младенцы с лиссэнцефалией могут иметь врожденные кардиологические проблемы органического характера в виде дефекта межпредсердной перегородки или сложного порока сердца с цианозом (тетрады Фалло). [24]
Последствия недостаточности постнатального развития в большинстве случаев приводят в летальному исходу в течение 24 месяцев после рождения.
Диагностика лиссэнцефалии
Диагностика начинается с физического осмотра ребенка, изучения анамнеза родителей и истории беременности и родов.
В период гестации могут потребоваться анализы внеклеточной ДНК плода, амниоцентез или взятие проб ворсинок хориона. [25] Подробнее см. – Пренатальная диагностика врожденных заболеваний
Для визуализации структур мозга и оценки их функций применяется инструментальная диагностика:
Во время беременности лиссэнцефалия на УЗИ плода после 20-21 недели может быть заподозрена при отсутствии теменно-затылочной и шпорной борозд и аномалии сильвиевой борозды головного мозга.
Дифференциальная диагностика
Проводится дифференциальная диагностика с другими синдромами врожденных церебральных пороков.
Существует более 20 типов лиссэнцефалии, большинство из которых подразделяются на 2 основные категории: классическая лиссэнцефалия (тип 1) и булыжная лиссэнцефалия (тип 2). Каждая категория имеет схожие клинические проявления, но разные генетические мутации. [27]
Обследование головного мозга при лизэнцефалии I типа показывает кору головного мозга с четырьмя слоями вместо шести, как у нормальных пациентов, тогда как при лизэнцефалии 2 типа кора головного мозга дезорганизована и выглядит бугристой или узловатой из-за полного смещения коры головного мозга скоплениями. корковых нейронов, разделенных глиомезенхимальной тканью. У пациентов также были аномалии мышц и глаз.
Микролисэнцефалия: это сочетание отсутствия нормальной складки коры головного мозга и аномально маленькой головы. Дети с обычной лиссэнцефалией при рождении имеют нормальный размер головы. У детей с уменьшенным размером головы при рождении обычно диагностируется микролисэнцефалия.
Также важно различать лизэнцефалию и полимикрогирию, которая представляет собой разные пороки развития мозга.