СОДЕРЖАНИЕ
Симптомы
Основные симптомы СПВ включают:
Гипертрофия: гипертрофия означает чрезмерный рост костей и мягких тканей. У пациентов с СПВ конечность зарастает, и обычно наблюдается гипертрофия пораженной конечности.
Капиллярные артериовенозные мальформации: нарушение сосудистой системы является причиной капиллярных мальформаций. Здесь капилляры увеличены и увеличивают приток крови к поверхности кожи. Из-за пороков развития капилляров на коже появляются многочисленные маленькие круглые, розовые или даже красные точки. У большинства пораженных людей эти пороки развития возникают на лице, руках и / или ногах. Пятна могут быть видны с самого рождения или появиться в детстве. Если пороки развития капилляров возникают сами по себе, это не представляет большой угрозы для жизни. Но когда они возникают в сочетании с AVF, это явный индикатор PWS и может быть серьезным в зависимости от серьезности пороков развития.
Аномальное кровотечение: некоторые кожные поражения легко кровоточат.
Периферический артериовенозный свищ: ненормальное сообщение между артерией и веной, которое является прямым результатом ненормального соединения или проводки между артерией и веной.
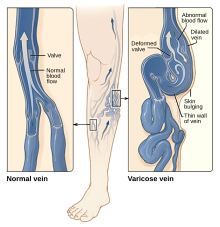
Варикозное расширение вен : увеличенные, опухшие и перекрученные вены.
Застойная сердечная недостаточность : это состояние, при котором снижается способность сердца удовлетворять потребности организма. Сердечный выброс уменьшаетсяа количество крови перекачивается не является адекватным достаточночтобы держать циркуляцию от тела и легких происходит.
Причины
Причины СПВ либо генетические, либо неизвестны. Некоторые случаи являются прямым результатом мутаций гена RASA1. И людей с RASA1 можно идентифицировать, потому что эта генетическая мутация всегда вызывает множественные пороки развития капилляров. PWS демонстрирует аутосомно-доминантный образец наследования. Это означает, что одной копии поврежденного или измененного гена достаточно, чтобы вызвать нарушение СПВ. В большинстве случаев СПВ возникает у людей, у которых в семейном анамнезе не было этого заболевания. В таких случаях мутация носит спорадический характер. А для пациентов с СПВ при отсутствии множественных капиллярных мутаций причины неизвестны.
По данным детской больницы Бостона, никакие известные продукты питания, лекарства или лекарства не могут вызвать СПВ во время беременности. PWS не передается от человека к человеку. Но он может передаваться по наследству и передаваться по наследству. PWS одинаково влияет как на мужчин, так и на женщин, и на данный момент расового преобладания не обнаружено.
Механизм
Мутации в гене RASA1 нарушают нормальное образование белка p120-RasGAP и приводят к нефункциональному белку. Белок больше не регулирует сигнальный путь RAS / MAPK. Однако, согласно домашнему справочнику NIH Genetics, до сих пор неясно, как именно нарушение образования белка p120-RasGAP приводит к сосудистым аномалиям и разрастанию конечностей. Но это известный факт, что каким-то образом белок p120-RasGAP имеет решающее значение для нормального развития сосудистой системы и ее сложной сети кровеносных сосудов, таких как артерии, вены и капилляры.
Основываясь на текущих знаниях, нарушение белка p120-RasGAP является причиной пороков развития кровеносных сосудов, которые, в свою очередь, приводят к разного рода проблемам, таким как чрезмерный рост конечностей, избыточный кровоток у поверхности кожи, что приводит к появлению винных пятен и может возникнуть даже сердечная недостаточность. Серьезность симптомов зависит от степени пороков развития.
Диагностика
Поставить правильный диагноз генетического и редкого заболевания часто бывает очень сложно. Таким образом, врачи и другие медицинские работники полагаются на историю болезни человека, тяжесть симптомов, физический осмотр и лабораторные тесты, чтобы поставить и подтвердить диагноз.
Обычно определенный набор симптомов, таких как капиллярная и артериовенозная мальформации, встречается вместе, и это используется для того, чтобы отличить СПВ от аналогичных состояний. Артериовенозные мальформации (АВМ) и артериовенозные свищи (АВФ) также вызываются мутациями RASA1. Следовательно, если все другие тесты (обсуждаемые ниже) не могут определить СПВ, что очень маловероятно, можно провести генетическое тестирование, такое как анализ последовательности и нацеленный на удаление / дупликацию генов, для выявления возможных мутаций гена RASA1.
МРТ: это сканирование с высоким разрешением, которое используется для определения степени гипертрофии или разрастания тканей. Это также может быть использовано для выявления других осложнений, которые могут возникнуть в результате гипертрофии.
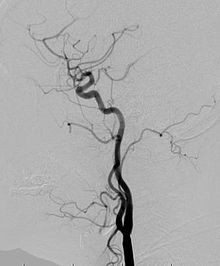
Ультразвук: это может быть необходимо для исследования сосудистой системы и определения количества крови, фактически протекающей через АВМ.
Компьютерная томография / компьютерная томография: это сканирование особенно полезно для исследования участков, пораженных СПВ, и для оценки костей разросшейся конечности.
Ангиограмма: также можно заказать ангиограмму, чтобы детально изучить кровеносные сосуды пораженной или разросшейся конечности. В этом тесте врач вводит краситель в кровеносные сосуды, который помогает увидеть, как кровеносные сосуды деформированы.
Эхокардиограмма: в зависимости от интенсивности синдрома PWS также может быть заказано эхо для проверки состояния сердца.
Дифференциальная диагностика
Профилактика
Другие варианты лечения включают эмболизацию и лазерную терапию. Эмболизация включает введение вещества, вводимого интервенционными радиологами, которое может помочь в устранении аномальных связей между артериями и венами. Согласно «Синдром Паркеса Вебера. Диагностические и лечебные парадигмы: систематический обзор», опубликованному в июле 2017 года, только эмболизация или в сочетании с хирургическим удалением артериовенозных мальформаций приводит к значительному клиническому улучшению. Лазерная терапия также может помочь облегчить капиллярные мальформации и ускорить процесс заживления кровоточащих поражений.
Прогноз
Недавние исследования
В другом обзоре, опубликованном в июле 2017 г. (обсуждаемом в разделе «Лечение и прогноз»), Banzic et al. обсудили клинические данные о том, что эмболизация действительно эффективна у пациентов с СПВ. Кроме того, эмболизация наряду с хирургической резекцией, направленной на артериовенозные мальформации, надежно приводит к значительным клиническим улучшениям.
Синдром Вебера

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Такое заболевание, как синдром Вебера, было впервые описано английским доктором Германом Дэвидом Вебером в XIX веке. Основными характеристиками синдрома являются: односторонний глазодвигательный паралич, гемиплегия и гемипарез, а также поражение лицевого и подъязычного нерва. Иногда заболевание осложняется гемианопсией.
Синдром Вебера – болезнь достаточно сложная и редкая, и представляет собой один из вариантов неврологической патологии из разряда педункулярных альтернирующих синдромов.
Код по МКБ-10
Эпидемиология
Эпидемиология синдрома Вебера не исследовалась.
 [1], [2], [3], [4]
[1], [2], [3], [4]
Причины синдрома Вебера
Появление заболевания связывают с патологическими изменениями, происходящими в непосредственной близости к ножкам мозга. Такие изменения могут быть результатом расстройства мозгового кровообращения (ишемия головного мозга), нарушения целостности сосудов мозга, опухолевых процессов.
Кроме того, развитие патологии может быть связано с локализованным давлением новообразования на ножки мозга, даже если опухоль расположена на некотором расстоянии от данной области.
[5], [6], [7]
Факторы риска
Можно выделить следующие факторы риска синдрома Вебера:
[8], [9]
Патогенез
Патогенез синдрома может заключаться:
[10], [11], [12], [13], [14], [15]
Симптомы синдрома Вебера
Первые признаки заболевания – это нарастающий паралич лицевой мускулатуры, мышц языка, рук и ног по центральному варианту. Клиническая симптоматика объясняется абсолютным или частичным обездвиживанием глазодвигательного нерва. Мышечное нарушение приводит к вынужденному отклонению глазного яблока в височную сторону. Выглядит это так, будто глаз «смотрит» в обратную сторону от пораженной стороны.
При одновременном поражении путей зрительной системы возникает гемианопсия – двусторонняя слепота половины поля зрения. У пациента наблюдается широкое косоглазие, зрительная функция падает, цвета и оттенки различаются с большим напряжением.
Кроме этого, могут обнаруживаться интенсивные и ритмичные движения по типу клонуса, вызванные толкательными мышечными сокращениями. Со временем состояние больного ухудшается: нарушается функция сгибания кисти на уровне защитного рефлекса.
Формы
Синдром Вебера относится к альтернирующим синдромам, суть которых состоит в функциональном расстройстве черепно-мозговых нервов со стороны повреждения, а также в расстройстве двигательной активности (в виде парезов и параличей), потере чувствительности (проводниковый вариант) и координации движений.
В зависимости от расположения патологического очага подобные синдромы подразделяют на следующие виды:
Синдром Вебера относят к педункулярным разновидностям заболевания.
[16], [17], [18], [19], [20]
Синдром Клиппеля-Треноне-Вебера
Синдром Клиппеля-Треноне-Вебера схож с описываемым нами синдромом Вебера лишь по названию. Суть же заболевания существенно отличается: патология связана с врожденным дефектом в сосудистой системе, который закладывается ещё в эмбриональном периоде.
Для болезни характерно появление на конечности невуса по типу телеангиоэктазии, на фоне варикозного расширения вен и венозно-артериальными анастомозами со стороны поражения. Нередки случаи развития парциального гигантизма пораженной ноги или (реже) руки. У некоторых пациентов обнаруживается искривление позвоночника, тазобедренный вывих, деформативные изменения суставов, стоп. Также видоизменяются сосуды зрительных органов, легких, почек.
Лечение патологии проводится оперативным путем.
Второе название синдрома Клиппеля-Треноне – это синдром Паркса-Вебера-Рубашова, или просто синдром Вебера-Рубашова.
[21], [22], [23], [24], [25]
Синдром Стерджа-Вебера-Краббе
Еще один наследственный синдром Стерджа-Вебера-Краббе характеризуется такими признаками, которые проявляются непосредственно после рождения малыша:
Лечение синдрома симптоматическое.
Иначе синдром называют энцефалотригеминальным ангиоматозом.
Синдром Вебера-Ослера
Точное название синдрома Вебера-Ослера – это заболевание Рандю-Вебера-Ослера.
Основой данной патологии является недостаток в трансмембранном белке эндоглине, который является ингредиентом рецепторной системы трансформирующего фактора роста β. Заболевание передается аутосомно-доминантным наследованием и характеризуется следующими симптомами:
Синдром проявляется уже в грудном возрасте, усугубляясь после наступления половой зрелости.
Диагностика синдрома Вебера
Диагностика синдрома Вебера может представлять определенные трудности. К сожалению, нет специфических методов, стопроцентно определяющих заболевание. Поэтому приходится использовать полный диагностический комплекс, для того чтобы правильно установить диагноз.
[26], [27], [28], [29]
Дифференциальная диагностика
Дифференциальная диагностика проводится с офтальмологическими заболеваниями, острым нарушением мозгового кровообращения, опухолевыми новообразованиями.
[30], [31], [32], [33], [34]
Лечение синдрома Вебера
Лечение синдрома Вебера должно быть направлено на устранение основной причины патологических изменений в зоне основания мозга. Поэтому направленность лечения – это терапия нарушений мозгового кровообращения, сосудистых нарушений, воспалительных процессов мозговых оболочек, удаление опухолевых новообразований, аневризмов и пр.
Могут быть назначены:
При необходимости врачи прибегают к хирургическому вмешательству – коррекции сосудистых и структурных нарушений.
В настоящее время одним из наиболее эффективных способов лечения альтернирующих синдромов любого происхождения считается пересадка стволовых клеток.
Стволовые клетки после трансплантации в мозг активируют восстановление тканей (в том числе и нервных тканей), что создает крайне благоприятные условия для лечения и регенерации поврежденных мозговых структур. После окончательного восстановления тканей мозга течение синдрома Вебера значительно облегчается.
Профилактика
Синдром Вебера не является самостоятельной патологией: как правило, это следствие или осложнение других заболеваний или повреждений, связанных с расстройством кровообращения в головном и спинном мозге. По этой причине профилактические мероприятия для предотвращения развития синдрома должны быть направлены на предупреждение различных нарушений внутримозгового кровообращения.
Какие рекомендации включает в себя подобная профилактика?
Кроме этого, врачи рекомендуют развивать в себе стрессоустойчивость, избегать конфликтных ситуаций. Все эти советы помогут сберечь нервную систему и предотвратить патологические изменения.
[35], [36], [37], [38]
Прогноз
Прогноз при своевременно оказанной медицинской помощи и при небольшой степени давления на ножки мозга может быть относительно благоприятным. Хуже, если поражение обширное, либо вызвано опухолевым процессом. В этом случае нарушенные мозговые функции могут не восстановиться.
Последствия синдрома Вебера могут быть различными:
Больной с признаками такого заболевания, как синдром Вебера, должен обязательно находиться под постоянным врачебным наблюдением. Даже при относительно стабильном состоянии, без видимого ухудшения, не следует терять бдительность: негативные последствия могут возникнуть и через время.
[39], [40]
Другие факоматозы, не классифицированные в других рубриках
Рубрика МКБ-10: Q85.8
Содержание
Определение и общие сведения [ править ]
Синонимы: множественная гамартома
Синдром Коудена является трудно распознаваемым и плохо диагностируемым генодерматозом. Характеризуется наличием множественных гамартом в различных тканях и повышенным риском развития злокачественных новообразований молочной железы, щитовидной железы, эндометрия, почек и колоректальной области. Если синдром Коудена ассоциирован с зародышевой мутацией PTEN, его относят в группу синдрома PTEN-гамартомы.
Распространенность неизвестна, но оценивается в 1/200000. Наследуется аутосомно доминантно.
Этиология и патогенез [ править ]
Клинические проявления [ править ]
Манифестация синдрома Коудена обычно происходят между второй и третьей декады жизни, но может появиться в любом возрасте. Патогномоничные дерматологические поражения (маленькие, цветные и множественные папулы на лица) могут быть первыми проявлениями болезни и наблюдаются у многих пациентов. Кромет того отмечаются папилломатозные папулы и акральный кератоз и проявления болезни Лермит-Дюкло. Возможны макроцефалия и дисморфизм лица, если они присутствуют, то наблюдаются с рождения. Часто развиваются злокачественные новообразования, такие как рак молочной железы (пожизненный риск 85%), эпителиальный рак щитовидной железы и рак эндометрия. Другие проявления, со стороны щитовидной железы: киста щитовидно-язычного протока, аденома; пищеварительного тракта: дивертикулит толстого кишечника, киста печени, гликогенный акантоз; мочеполовой системы: функциональные нарушения менструального цикла, овариальные тератомы; скелета: кисты в костях; молочной железы: фиброзно-кистозная болезнь, аномалии соска и ареолы; нервной системы: менингиомы, задержка развития, аутизм), все они имеют весьма разнообразный возраст начала.
Другие факоматозы, не классифицированные в других рубриках: Диагностика [ править ]
Диагноз формулируется, если у пациента отмечаются патогномоничные поражения кожи, два и более основных, один основной и 3 или более незначительных, 4 или более незначительных критериев патологии. К настоящему времени создана скориговая система для взрослых и отдельную для педиатрической практики. Обнаружение мутаций PTEN или других причинных генов подтверждает диагноз.
Дифференциальный диагноз [ править ]
Дифференциальный диагноз проводят с синдромом ювенильного полипоза, синдромом Пейтца-Егерса, синдромом Берта-Хогга-Дюбе, синдромом Горлина-Гольтца и нейрофиброматозом 1-го типа.
Другие факоматозы, не классифицированные в других рубриках: Лечение [ править ]
После того, как идентифицирована мутация PTEN, принципы наблюдения должны строго соблюдаться: регулярные УЗИ щитовидной железы с возраста 18 лет, колоноскопия и визуализация почек с возраста 35-40 лет. Женщины должны выполнять ежемесячно самопроверки груди и ежегодные скрининги молочной железы, а также трансвагинальное УЗИ или биопсию эндометрия, начиная с возраста 30 лет.
Своевременная постановка диагноза, особенно генетическая, приводит к хорошим прогнозом. При поздних стадиях рака неблагоприятный исход является распространенным явлением.
Профилактика [ править ]
Прочее [ править ]
Синонимы: энцефалофациальный ангиоматоз, синдром Стерджа-Вебера-Краббе, синдром Штурге-Вебера
Определение и общие сведения
Синдром Стерджа-Вебера является редкой врожденной нейро-кожной патологией. Синдром Штурге-Вебера характеризуется наличием аномалий капилляров на лице (ангиомы) и/или аномалий головного мозга и ипсилатеральных сосудистых пороков развития глаза.
Распространенность при рождаемости в Европе оценивается на уровне около 1/20000 и 1/50000. Заболевание носит спорадический характер.
Описали заболевание в 1879 г. английские врачи W.H. Sturge (1850-1919) и H.D. Weber (1823-1918).
Этиология и патогенез
Аномалии капилляров на лице (ангиомы) проявляются портвейновым невусом (пламенеющий невус), который, как правило, присутствует с рождения и расположен на лбу или на верхнем века, на одной или обеих сторонах лица. Иногда портвейновый невус может охватывать верхнюю и нижнюю челюсти, а в некоторых случаях может распространяться на туловище и конечностях. Гипертрофия костей и мягких тканей могут сопутствовать развитию невуса, что приводит к развитию нарушений зрения, слуха, глотания и артикуляции. В редких случаях портвейновый невусом отсутствует.
Поражение глаз может произойти в любом возрасте, но, как правило, наблюдается в детском или в подростковом возрасте. Более, чем у 50% пациентов развивается глаукома на той же самой стороне лица, где распологается невус, которая может приводить к атрофии зрительного нерва и слепоте.
Церебральные сосудистые нарушения проявляются обычно лептоменингиальным ангиоматозом с первого года жизни. Возникают очаговые или сложные парциальные судорожные припадки. Мигрень и инсульт-подобные эпизоды также очень распространены. С прогрессированием заболевания, и, в зависимости от тяжести приступов, у пациентов может развиться гемипарез, гемиплегия и различной степени умственная отсталость. Менее распространенные симптомы включают в себя повышенный риск развития дефицита гормона роста.
В зависимости от размеров невуса, риск развития синдрома Стерджа-Вебера у детей раннего возраста колеблется в пределах 15-40%. Диагноз подтверждается с помощью рентгенографии, КТ или МРТ с контрастом, обнаруживая ипсилатеральную церебральную гемиатрофию, корковые кальцификации и лептоменингиальной ангиоматоз. Используют также измерения церебрального кровотока и позитронно-эмиссионную томографию.
Дородовая диагностика возможна.
Главной диагностической проблемой является исключение случаев изолированного портвейнового невуса.
Прогноз зависит от тяжести эпилептических кризов, которые могут приводить к различной степени психомоторной регрессии и умственной отсталости.
Синонимы: семейный церебеллороретинальный ангиоматоз, синдром Гиппеля-Линдау
Определение и общие сведения
Болезнь Гиппеля-Линдау представляет собой синдром наследственной предрасположенности к злокачественным образованиям. Наиболее часто развиваются опухоли сетчатки, мозжечка, гемангиобластома спинного мозга, почечно-клеточный рак и феохромоцитома.
Этиология и патогенез
Гемангиобластомы сетчатки (множественный и двусторонние примерно в 50% случаев) являются наиболее распространенной причиной обращения при болезни Гиппеля-Линдау. Они, как правило, протекают бессимптомно, но могут вызывать отслоение сетчатки, макулярный отек, глаукому и потери зрения. Гемангиобластомы ЦНС обнаруживаются впервые примерно у 40% пациентов, а в целом наблюдаются у 60-80% больных. Чаще всего они расположены в мозжечке, но встречаются и в стволе головного мозга и спинном мозге. Они являются доброкачественными, но вызывают соответствующую симптоматику, вследствии компрессии смежной с ней нервной ткани. При локализации в мозжечке они чаще всего вызывают повышение внутричерепного давления, головные боли, рвоту и атаксию. Очень распространены множественные кисты почек, пожизненный риск возникновения почечно-клеточного рака очень высока (70%). Некоторые пациенты имеют феохромоцитомы, которые могут быть бессимптомными, но может приводить к развитию гипертонии. Могут возникать эпидидимальные кисты и цистоаденомамы (60% пациентов мужского пола), а также множественные кисты поджелудочной железы (большинство пациентов), но не-секреторные опухоли островковых клеток поджелудочной железы встречаются только у около 10% больных. Опухоли эндолимфатического мешка также были обнаруживаются у до 10% пациентов и может приводить к потере слуха. Параганглиомы головы и шеи встречаются редко (0,5%). Средний возраст на момент постановки диагноза опухолей при болезни Гиппеля-Линдау значительно моложе, чем в спорадических случаях. Отмечена внутрисемейных изменчивость.
Дифференциальный диагноз включают множественную эндокринную неоплазию, нейрофиброматоз, поликистоз почек, туберозный склероз, синдром Берта-Хогга-Дюбе и синдром наследственной феохромоцитомы-параганглиомы, ассоциированную с мутацией субъединиц сукцинатдегидрогеназы (SDHB, SDHC и SDHD).
Лечение требует скоординированного междисциплинарного подхода. Хирургия является основой лечения опухолей. Ведение должно включать пожизненное наблюдение (офтальмологическое, сканирование МРТ головного мозга и брюшной полости, лабораторные исследования). Родственники в группе риска должны быть введены в программу скрининга с детства, если болезнь Гиппеля-Линдау не исключается в ходе молекулярно-генетического тестирования.
Прогноз зависит от риска возникновение множественных опухолей. Почечно-клеточный рак является основной причиной смерти, затем гемангиобластомы ЦНС. Средняя продолжительность, по оценкам, составляет 50 лет. Тем не менее, регулярное наблюдение, раннее выявление и лечение опухолей в настоящее время снижается заболеваемость и смертность.
Синонимы: множественные гамартоматозные полипы кишечника
Определение и общие сведения
Синдром Пейтца-Егерса является наследственным заболеванием желудочно-кишечного тракта. Характеризуется развитием характерных гамартоматозные полипов в ЖКТ, а также кожной пигментацией. Синдром Пейтца-Егерса несет значительно повышенный риск развития желудочно-кишечных и экстра-интестинальных злокачественных опухолей.
Оценки распространенности в диапазоне от 1/25000 до 1/300 000 рождений в Соединенных Штатах. Синдром Пейтца-Егерса наследуется по аутосомно-доминантному типу.
Этиология и патогенез
Синдром Пейтца-Егерса вызван зародышевой мутацией гена-супрессоре опухоли STK11 (19p13.3). Мутации в этом гене встречаются более чем у 80% семей. Тем не менее отсутствуют четкие корреляции генотип/фенотип. Одно исследование показало, что люди с миссенс-мутацией, имели значительно позднее время наступления первой полипэктомией и других симптомов, по сравнению с лицами с мутацией по типу делеции или отсутствием обнаруживаемой мутации. Однако в другом исследовании, генотип-фенотипические корреляции не наблюдалось.
Несмотря на высокую изменчивость между семьями, характерные для синдрома Пейтца-Егерса полипы обычно возникают в детском и юношеском возрасте, но наиболее часто в течение первых 10 лет жизни. Гамартоматозные полипы могут возникнуть в любом участке желудочно-кишечного тракта, но наиболее часто в тонком кишечнике. Другие локализации включают в себя желудок, толстый кишечник, полость носа (ноздри) и редко почечные лоханки, мочевой пузырь и легкие. Хотя эти полипы доброкачественные, они могут приводить к осложнениям, в том числе к кишечной непроходимости, выпадению прямой кишки, а также тяжелым желудочно-кишечным кровотечениям со вторичной анемией и непроходимостью. Могут также обнаруживаться аденомы.
В младенчестве или детстве, у пациентов развиваются макулы вокруг рта, глаз, ноздрей, в перианальной области и на слизистой оболочки полости рта, от темно-синего до темно-коричневого цвета. Гиперпигментация также могут быть обнаруживаться на пальцах рук и ног. Эти симатомы могут исчезать в подростковом и взрослом возрасте, но, как правило, сохраняются в слизистой оболочки полости рта и могут вызывать психологический стресс у пациентов.
Интестинальные и экстра-интестинальные злокачественные опухоли в основном наблюдаются у взрослых пациентов и включают в себя рак прямой кишки и рак желудка (по оценкам, прижизненный риск 15% в 50 лет, и 57% в 70 лет), рак поджелудочной железы (прижизненный риск 5% в 50 лет и 17% в 70 лет), а также рак груди и яичников у женщин (прижизненный риск 8% в 40 лет, 32% в 60 лет). У пациенток может также развиться злокачественная аденома шейки матки, и, как правило, доброкачественные двусторонние мультифокальные опухоли полового тяжа с кольцевыми канальцами. Мужчины имеют более высокий риск развития крупно-клеткочной обызвествляющейся опухоли из клеток Сертоли.
Пренатальная диагностика доступна при условии, что мутация, вызывающие болезнь была обнаружена в семье.
Рутинные методы лечения используюь для резекции полипов, терапии инвагинации и злокачественных опухолей. Цель наблюдения заключается в снижении тяжести осложнений у молодых пациентов, а также для мониторинга злокачественных опухолей у пациентов пожилого возраста.
Прогноз зависит от тяжести осложнений и от развитие злокачественных опухолей.
Линейный невус сальных желез
Синонимы: невус сальных желез Ядассона, синдром Соломона, синдром Шиммепеннинга
Определение и общие сведения
Линейный невус сальных желез характеризуется наличием больших сальных невусов, обычно появляющихся на лице или на коже головы, которым сопутствует широкий спектр нарушений, которые могут затронуть любую систему организма, включая центральную нервную систему (опухоли головного мозга, гемимегалэнцефалия и увеличение боковых желудочков).
Заболеваемость невусом сальных желез при рождении оценивается в 1 на 1000, но распространенность этих поражений в сочетании с другими аномалиями примерно 1:10 000.
Этиология и патогенез
Линейный невус сальных желез является спорадическим заболеванием. Предполагается, что синдромы эпидермальных невусов являются результатом генетического мозаицизма с участием доминантного гена.
Основными неврологическими симтомами являются судороги (до 75% пациентов) и интеллектуальный дефицит (до 60% пациентов). Синдром может также затрагивать многие другие системы организма, включая сердечно-сосудистую (коарктация аорты), скелетную (локальная фиброзная дисплазия черепа, скелетная гипоплазия, образование костных структур, сколиоз и кифосколиоз, витамин Д-резистентный рахит и гипофосфатемия), органы зрения (косоглазие, аномалии сетчатки, колобома, катаракта, васкуляризация роговицы и окулярные гемангиомы) и мочеполовую систему (подковообразная почка).
Из-за возможности полиорганного поражения все дети с подозрением на синдром линейного невуса сальных желез должны проходить мультисистемное обследование с помощью КТ и, если возможно, МРТ-сканирование мозга, электроэнцефалограммы при эпилепсии, биопсии кожи и офтальмологических исследований.
Дифференциальная диагностика должна включать синдром комедонового невуса, синдром невуса Беккера, пигментнокератотический факоматоз, синдром Протея и синдром CHILD.
Заболевание обычно бессимптомно, но из-за его косметического воздействия и злокачественного потенциала может быть рекомендовано профилактическое удаление поражения (предпочтительно до наступления половой зрелости, а иногда даже в младенчестве или раннем детстве). Нейровизуализация по клиническим показаниям.
Прогноз зависит от тяжести клинических проявлений и от степени вовлеченности систем организма.
Синдром эпидермального невуса
Синонимы: синдром эпидермальной гамартомы
Около 50% пациентов имеют неврологические аномалии, которые включают умственную отсталость и эпилепсию, спастический парез, церебральные сосудистые мальформации, атрофию коры, увеличение боковых желудочков. Около трети пациентов могут иметь офтальмологические аномалии: колобома века, радужной оболочки и сетчатки, конъюнктивальные липодермоиды и хористомы, кортикальная слепота, микро-, макро- или анофтальмия, помутнение роговицы и катаракта. Могут присутствовать скелетные аномалии и другие некожные проявления.
Идеальной медицинской терапии кожных поражений не существует. Повреждения кожи могут поддаваться хирургическому вмешательству. Воспалительный линейный бородавчатый эпидермальный невус иногда реагирует на лазеротерапию или аналоги витамина D. Были опробованы салициловая кислота, топические и системные ретиноиды, смягчающие средства, дермабразия и криотерапия.
Сопутствующие дефекты скелета и аномалии глаза могут быть скорректированы хирургически. Эпилепсию требует назначения соответствующей терапии.
Определение и общие сведения
Для пигментоваскулярного факоматоза было предложено множество систем классификации, в основном в зависимости от типа пигментного поражения. Около половины пациентов с пигментоваскулярным факоматозом имеют разноообразные системные проявления.
Этиология и патогенез
Пигментоваскулярный факоматоз не наследуется, но, как полагают, вызывается генетическим явлением, называемым твин-споттинг (twin spotting). Предполагается, что происходит изменения расположения небольшого фрагмента генетического материала в развивающемся эмбрионе. Из-за этого изменения некоторые клетки тела ребенка несут две копии рецессивных генных мутаций, в то время как большинство клеток тела несут только одну.
Характерные симптомы пигментоваскулярного факоматоза включают портвейновый невус (пламенеющий невус) и пигментные поражения, которые часто обширны и могут затрагивать несколько областей тела, включая лицо.
Сопутствующие пигментных поражений включают в себя: меланоцитарные невусы, эпидермальные невусы, анемичный невус, пятна “кофе с молоком”, монгольское пятно, невус Ота, невус Ито, шпилюс-невус.
Около половины пациентов обнаруживают системное участие: окулярный меланоз (сине-серая пигментация склер). Окулярный меланоз часто отмечается совместно с невусом Отв и может поражать один или оба глаза. Осложнениями невуса Оты являются глаукома и меланома. Другие заболевания органа зрения включают в себя раздичные патологии радужки.
У части пациентов наблюдается синдром Стерджа-Вебера или синдром Клиппеля-Треноне.
При отсутствии системных проявлений лечение проводят только в косметических целях.
Источники (ссылки) [ править ]
Fernández-Guarino M, Boixeda P, de Las Heras E, Aboin S, García-Millán C, Olasolo PJ. Phakomatosis pigmentovascularis: Clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008 Jan